Nous étudions les bases épigénétiques de l’identité cellulaire chez les mammifères.
La différenciation d’une cellule en une cellule spécialisée est un processus complexe nécessitant l’action coordonnée de multiples niveaux de régulation pour assurer l’acquisition d’un nouveau programme d’expression génique. Pourtant, les mécanismes orchestrant ce processus, ainsi que les causes et conséquences de leurs altérations, restent encore largement méconnus.
Nous explorons cette question en analysant les mécanismes qui coordonnent les différentes régulations épigénétiques lors de l’acquisition d’une identité neurale, ainsi que leur dérégulation dans un contexte pathologique.
Pour cela, nous utilisons l’empreinte parentale et le gliome malin comme modèles d’étude.
Financeurs



Recherche
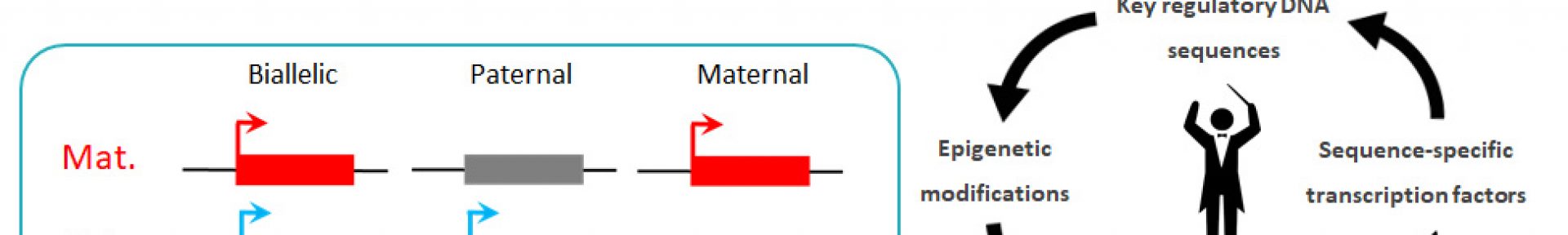
L’empreinte parentale : de la régulation à la fonction
Régulation des loci d’ empreinte dans les lignées neurales (Contacts : F. Court, P. Arnaud)
L’empreinte génomique limite l’expression de près de 150 gènes de mammifères à un seul allèle parental. Ces gènes jouent un rôle clé dans le contrôle de la croissance et le fonctionnement du cerveau. Au niveau moléculaire, ils sont organisés en une vingtaine de domaines génomiques, chacun régulé par une région de contrôle de l’empreinte (ICR) et l’action coordonnée de multiples niveaux de régulation. Cependant, les mécanismes qui orchestrent cette régulation multi-échelle et permettent à l’ICR de contrôler à distance l’expression monoallélique des gènes lors de l’acquisition de l’identité cellulaire restent mal compris. Notre objectif est d’identifier ces mécanismes et les acteurs clés impliqués dans l’acquisition de l’identité neurale.
Pour y parvenir, nous devons évaluer simultanément la dynamique de plusieurs niveaux de régulation au cours de l’acquisition de l’identité cellulaire. À cette fin, nous avons développé une approche intégrée qui combine des études expérimentales multi-échelles avec des analyses fonctionnelles en utilisant des systèmes de corticogenèse in vitro 2D et 3D (organoïdes) qui récapitulent les modèles d’empreinte observés dans le cerveau précoce (Bouschet et al., Cereb Cortex, 2017) et postnatal, respectivement. Ce système réduit à l’animal repose sur la différenciation contrôlée de cellules souches embryonnaires (ES) murines hybrides réciproques, permettant l’étude dynamique et allélique des signatures moléculaires.
Notre stratégie intègre des analyses moléculaires complètes (méthylation de l’ADN, expression des gènes, modifications des histones et facteurs de régulation), des analyses de conformation de la chromatine en 3D et des analyses bioinformatiques. Cela nous a permis de générer une carte à haute résolution offrant une vue intégrative, allélique et dynamique des signatures moléculaires linéaires et de l’organisation de la chromatine en 3D associées aux régions ICR pendant la formation de la lignée neuronale murine. Ce cadre nous permet de disséquer les mécanismes de régulation de multiples loci d’empreinte, que nous testons fonctionnellement à l’aide d’approches basées sur CRISPR.

Fonction des loci d’empreinte : le locus H13/Mcts2 (Contacts : B. Montibus, P. Arnaud)
Les perturbations de la régulation transcriptionnelle de l’empreinte génomique, y compris les altérations de la production d’isoformes spécifiques d’allèles, peuvent entraîner des troubles de l’empreinte, un groupe de maladies congénitales rares affectant la croissance et le développement neurologique. L’identification des causes génétiques et des mécanismes physiopathologiques sous-jacents aux troubles de l’empreinte reste un défi majeur.
Un exemple frappant est le syndrome de Mulchandani-Bhoj-Conlin (MBCS), une maladie caractérisée par un retard de croissance fœtale et postnatale, une petite taille, et un retard de développement. Des données indirectes suggèrent qu’une expression dérégulée du locus HM13/Mcts2 contribue au MBCS. Pour vérifier cette hypothèse, notre projet vise à élucider les mécanismes de régulation et les rôles fonctionnels du locus H13/Mcts2 afin d’évaluer son impact sur la pathogenèse du MBCS.Pour ce faire, nous utilisons une combinaison d’analyses phénotypiques et d’études moléculaires au niveau transcriptionnel et translationnel dans des modèles murins et des lignées cellulaires. Notre approche intègre des études descriptives et fonctionnelles pour découvrir les rôles de l’expression des allèles spécifiques et des isoformes au cours du développement et les conséquences de leur dérégulation dans la maladie.

Le Gliome : étude des transcrits chimères issues d’élément LINE L1
( contacts: C. Vaurs-Barriere; P Arnaud)
Ce projet a été initié pour identifier les mécanismes qui coordonnent les différents niveaux de régulation épigénétiques en recherchant les causes et les conséquences de leur altération dans le gliome malin, l’un des types le plus répandu de tumeurs cérébrales. Des collaborations avec des équipes cliniques nous permettent de mener cette étude sur les échantillons de la tumorothéque Auvergne Gliome et des lignées de cellules initiatrices de glioblastomes (CIG). Une étude moléculaire exhaustive, nous a permis de révéler que la majorité des défauts transcriptionnel dans les gliomes ne sont pas liés à des défauts de méthylation ADN mais plutôt dus à une altération dans le contrôle de H3K27me3 (ici). La recherche des causes de cette altération nous conduit à étudier plus en détail la (dé) régulation gènes HOX, pour lesquels nous avons établis que l’altération est une signature des gliomes les plus agressifs.
Pour identifier de nouveaux candidats nous avons entrepris, dans une approche parallèle, d’explorer le lien entre dérégulation des éléments transposables et altération de H3K27me3 dans les cellules cancéreuses. Notre hypothèse originale est que la transcription antisens depuis les séquences répétées L1 peut induire l’expression aberrante de gènes adjacents, sous la forme de transcrit dits chimères ou LCT (LINE-1 chimeric transcripts), qui vont eux même influencer la dynamique de la marque H3K27me3. Nous avons précédemment développé l’outil bio-informatique CLIFinder afin d’identifier ces transcrits chimères à partir de données RNA seq (ici). Fort de cet outil, nous avons, pour la première fois, établi le panorama des LCTs dans les gliomes agressifs et entrepris de caractériser, dans les CIGs, les gènes ainsi altérés.

Membres









Publications
Mechanisms controlling genomic imprinting and their dysregulation in human imprinting disorders.
Publié le 15 Avr 2026 dans Annales d'endocrinologie , vol. 87 - pp 102552
Juven A, Vaurs-Barrière C , Court F , Montibus B , Arnaud P
The superpowers of imprinting control regions.
Publié le 10 Déc 2025 dans Genome research , vol. 36 - pp 1-19
PIK3R1 and G0S2 are human placenta-specific imprinted genes associated with germline-inherited maternal DNA methylation.
Publié le 01 Déc 2025 dans Epigenetics , vol. 20 - pp 2523191
Daskeviciute D, Sainty B, Chappell-Maor L, Bone C, Russell S, Iglesias-Platas I, Arnaud P , Monteagudo-Sánchez A, Greenberg MVC, Chen K, Manerao-Azua A, Perez de Nanclares G, Lartey J, Monk D
Non-canonical imprinting, manifesting as post-fertilization placenta-specific parent-of-origin dependent methylation, is not conserved in humans.
Publié le 17 Jan 2025 dans Human molecular genetics
Daskeviciute D, Chappell-Maor L, Sainty B, Arnaud P , Iglesias-Platas I, Simon C, Okae H, Arima T, Vassena R, Lartey J, Monk D
Biallelic non-productive enhancer-promoter interactions precede imprinted expression of Kcnk9 during mouse neural commitment.
Publié le 30 Jan 2024 dans HGG advances , vol. 5 - pp 100271
Rengifo Rojas C , Cercy J , Perillous S , Gonthier-Guéret C , Montibus B , Maupetit-Méhouas S, Espinadel A , Dupré M , Hong CC, Hata K, Nakabayashi K, Plagge A, Bouschet T, Arnaud P , Vaillant I , Court F
Variable allelic expression of imprinted genes at the Peg13, Trappc9, Ago2 cluster in single neural cells.
Publié le 12 Oct 2022 dans Frontiers in cell and developmental biology , vol. 10 - pp 1022422
Claxton M, Pulix M, Seah MKY, Bernardo R, Zhou P, Aljuraysi S, Liloglou T, Arnaud P , Kelsey G, Messerschmidt DM, Plagge A
L1 chimeric transcripts are expressed in healthy brain and their deregulation in glioma follows that of their host locus.
Publié le 17 Août 2022 dans Human molecular genetics , vol. 31 - pp 2606-2622
Pinson ME , Court F , Masson A , Renaud Y , Fantini A , Bacoeur-Ouzillou O, Barriere M, Pereira B, Guichet PO, Chautard E, Karayan-Tapon L, Verrelle P, Arnaud P , Vaurs-Barrière C
The Long Non-Coding RNA HOXA-AS2 Promotes Proliferation of Glioma Stem Cells and Modulates Their Inflammation Pathway Mainly through Post-Transcriptional Regulation.
Publié le 25 Avr 2022 dans International journal of molecular sciences , vol. 23
Le Boiteux E , Guichet PO, Masliantsev K, Montibus B , Vaurs-Barriere C, Gonthier-Gueret C, Chautard E, Verrelle P, Karayan-Tapon L, Fogli A , Court F , Arnaud P
Widespread overexpression from the four DNA hypermethylated HOX clusters in aggressive (IDHwt) glioma is associated with H3K27me3 depletion and alternative promoter usage.
Publié le 30 Août 2021 dans Molecular oncology , vol. 15 - pp 1995-2010
Le Boiteux E , Court F , Guichet PO, Vaurs-Barrière C , Vaillant I , Chautard E, Verrelle P, Costa BM, Karayan-Tapon L, Fogli A , Arnaud P
TET3 controls the expression of the H3K27me3 demethylase Kdm6b during neural commitment.
Publié le 30 Jan 2021 dans Cellular and molecular life sciences : CMLS , vol. 78 - pp 757-768
Montibus B , Cercy J , Bouschet T, Charras A, Maupetit-Méhouas S , Nury D , Gonthier-Guéret C , Chauveau S , Allegre N , Chariau C, Hong CC, Vaillant I , Marques CJ, Court F , Arnaud P
HOX gene cluster (de)regulation in brain: from neurodevelopment to malignant glial tumours.
Publié le 30 Oct 2020 dans Cellular and molecular life sciences : CMLS , vol. 77 - pp 3797-3821
Gonçalves CS, Le Boiteux E , Arnaud P , Costa BM
Transcriptional alterations in glioma result primarily from DNA methylation-independent mechanisms.
Publié le 30 Oct 2019 dans Genome research , vol. 29 - pp 1605-1621
Court F , Le Boiteux E , Fogli A , Müller-Barthélémy M, Vaurs-Barrière C , Chautard E, Pereira B, Biau J, Kemeny JL, Khalil T, Karayan-Tapon L, Verrelle P, Arnaud P
Childhood Ataxia with Central Nervous System Hypomyelination / Vanishing White Matter.
Publié le 04 Avr 2019
Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, van der Knaap MS, Fogli A , Boespflug-Tanguy O, Abbink TEM, Schiffmann R
High-salt-recovered sequences are associated with the active chromosomal compartment and with large ribonucleoprotein complexes including nuclear bodies.
Publié le 30 Nov 2018 dans Genome research , vol. 28 - pp 1733-1746
Baudement MO, Cournac A, Court F , Seveno M, Parrinello H, Reynes C, Sabatier R, Bouschet T, Yi Z, Sallis S, Tancelin M, Rebouissou C, Cathala G, Lesne A, Mozziconacci J, Journot L, Forné T
Oxidative stress and mitochondrial dynamics malfunction are linked in Pelizaeus-Merzbacher disease.
Publié le 30 Sep 2018 dans Brain pathology (Zurich, Switzerland) , vol. 28 - pp 611-630
Ruiz M, Bégou M, Launay N, Ranea-Robles P, Bianchi P, López-Erauskin J, Morató L, Guilera C, Petit B, Vaurs-Barriere C , Guéret-Gonthier C, Bonnet-Dupeyron MN, Fourcade S, Auwerx J, Boespflug-Tanguy O, Pujol A
DNA methylation profiling reveals a pathological signature that contributes to transcriptional defects of CD34(+) CD15(-) cells in early chronic-phase chronic myeloid leukemia.
Publié le 30 Juin 2018 dans Molecular oncology , vol. 12 - pp 814-829
Maupetit-Mehouas S , Court F , Bourgne C, Guerci-Bresler A, Cony-Makhoul P, Johnson H, Etienne G, Rousselot P, Guyotat D, Janel A, Hermet E, Saugues S, Berger J, Arnaud P , Berger MG
The long non-coding RNA HOTAIR is transcriptionally activated by HOXA9 and is an independent prognostic marker in patients with malignant glioma.
Publié le 20 Mar 2018 dans Oncotarget , vol. 9 - pp 15740-15756
Xavier-Magalhães A, Gonçalves CS, Fogli A , Lourenço T, Pojo M, Pereira B, Rocha M, Lopes MC, Crespo I, Rebelo O, Tão H, Lima J, Moreira R, Pinto AA, Jones C, Reis RM, Costello JF, Arnaud P , Sousa N, Costa BM
CLIFinder: identification of LINE-1 chimeric transcripts in RNA-seq data.
Publié le 15 Fév 2018 dans Bioinformatics (Oxford, England) , vol. 34 - pp 688-690
Pinson ME , Pogorelcnik R , Court F , Arnaud P , Vaurs-Barrière C
Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains.
Publié le 01 Jan 2018 dans Epigenetics , vol. 13 - pp 117-121
Monk D, Morales J, den Dunnen JT, Russo S, Court F , Prawitt D, Eggermann T, Beygo J, Buiting K, Tümer Z, Nomenclature group of the European Network for Human Congenital Imprinting Disorders
Detection of the alternative lengthening of telomeres pathway in malignant gliomas for improved molecular diagnosis.
Publié le 30 Nov 2017 dans Journal of neuro-oncology , vol. 135 - pp 381-390
Fogli A , Demattei MV, Corset L, Vaurs-Barrière C , Chautard E, Biau J, Kémény JL, Godfraind C, Pereira B, Khalil T, Grandin N , Arnaud P , Charbonneau M , Verrelle P
Maternal mutations of FOXF1 cause alveolar capillary dysplasia despite not being imprinted.
Publié le 30 Juin 2017 dans Human mutation , vol. 38 - pp 615-620
Alsina Casanova M, Monteagudo-Sánchez A, Rodiguez Guerineau L, Court F , Gazquez Serrano I, Martorell L, Rovira Zurriaga C, Moore GE, Ishida M, Castañon M, Moliner Calderon E, Monk D, Moreno Hernando J
In Vitro Corticogenesis from Embryonic Stem Cells Recapitulates the In Vivo Epigenetic Control of Imprinted Gene Expression.
Publié le 01 Mar 2017 dans Cerebral cortex (New York, N.Y. : 1991) , vol. 27 - pp 2418-2433
Bouschet T, Dubois E, Reynès C, Kota SK, Rialle S, Maupetit-Méhouas S , Pezet M, Le Digarcher A, Nidelet S, Demolombe V, Cavelier P, Meusnier C, Maurizy C, Sabatier R, Feil R, Arnaud P , Journot L, Varrault A
DNA methylation profiling identifies PTRF/Cavin-1 as a novel tumor suppressor in Ewing sarcoma when co-expressed with caveolin-1.
Publié le 01 Fév 2017 dans Cancer letters , vol. 386 - pp 196-207
Huertas-Martínez J, Court F , Rello-Varona S, Herrero-Martín D, Almacellas-Rabaiget O, Sáinz-Jaspeado M, Garcia-Monclús S, Lagares-Tena L, Buj R, Hontecillas-Prieto L, Sastre A, Azorin D, Sanjuan X, López-Alemany R, Moran S, Roma J, Gallego S, Mora J, García Del Muro X, Giangrande PH, Peinado MA, Alonso J, de Alava E, Monk D, Esteller M, Tirado OM
An annotated list of bivalent chromatin regions in human ES cells: a new tool for cancer epigenetic research.
Publié le 17 Jan 2017 dans Oncotarget , vol. 8 - pp 4110-4124
Quantitative Analysis of Intra-chromosomal Contacts: The 3C-qPCR Method.
Publié le 01 Jan 2017 dans Methods in molecular biology (Clifton, N.J.) , vol. 1589 - pp 75-88
Ea V, Court F , Forné T
Human Oocyte-Derived Methylation Differences Persist in the Placenta Revealing Widespread Transient Imprinting.
Publié le 30 Nov 2016 dans PLoS genetics , vol. 12 - pp e1006427
Sanchez-Delgado M, Court F , Vidal E, Medrano J, Monteagudo-Sánchez A, Martin-Trujillo A, Tayama C, Iglesias-Platas I, Kondova I, Bontrop R, Poo-Llanillo ME, Marques-Bonet T, Nakabayashi K, Simón C, Monk D
The tumoral A genotype of the MGMT rs34180180 single-nucleotide polymorphism in aggressive gliomas is associated with shorter patients’ survival.
Publié le 01 Mar 2016 dans Carcinogenesis , vol. 37 - pp 169-176
Fogli A , Chautard E, Vaurs-Barrière C , Pereira B, Müller-Barthélémy M, Court F , Biau J, Pinto AA, Kémény JL, Khalil T, Karayan-Tapon L, Verrelle P, Costa BM, Arnaud P
Imprinting control regions (ICRs) are marked by mono-allelic bivalent chromatin when transcriptionally inactive.
Publié le 29 Jan 2016 dans Nucleic acids research , vol. 44 - pp 621-35
Maupetit-Méhouas S , Montibus B , Nury D , Tayama C, Wassef M, Kota SK, Fogli A , Cerqueira Campos F , Hata K, Feil R, Margueron R, Nakabayashi K, Court F , Arnaud P
Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape.
Publié le 25 Sep 2015 dans Genome biology , vol. 16 - pp 209
Veselovska L, Smallwood SA, Saadeh H, Stewart KR, Krueger F, Maupetit-Méhouas S , Arnaud P , Tomizawa S, Andrews S, Kelsey G
ICR noncoding RNA expression controls imprinting and DNA replication at the Dlk1-Dio3 domain.
Publié le 13 Oct 2014 dans Developmental cell , vol. 31 - pp 19-33
Kota SK, Llères D, Bouschet T, Hirasawa R, Marchand A, Begon-Pescia C, Sanli I, Arnaud P , Journot L, Girardot M, Feil R
Variable maternal methylation overlapping the nc886/vtRNA2-1 locus is locked between hypermethylated repeats and is frequently altered in cancer.
Publié le 30 Mai 2014 dans Epigenetics , vol. 9 - pp 783-90
Romanelli V, Nakabayashi K, Vizoso M, Moran S, Iglesias-Platas I, Sugahara N, Simón C, Hata K, Esteller M, Court F , Monk D
Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment.
Publié le 30 Avr 2014 dans Genome research , vol. 24 - pp 554-69
Court F , Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, Simón C, Moore H, Harness JV, Keirstead H, Sanchez-Mut JV, Kaneki E, Lapunzina P, Soejima H, Wake N, Esteller M, Ogata T, Hata K, Nakabayashi K, Monk D
The PEG13-DMR and brain-specific enhancers dictate imprinted expression within the 8q24 intellectual disability risk locus.
Publié le 25 Mar 2014 dans Epigenetics & chromatin , vol. 7 - pp 5
Court F , Camprubi C, Garcia CV, Guillaumet-Adkins A, Sparago A, Seruggia D, Sandoval J, Esteller M, Martin-Trujillo A, Riccio A, Montoliu L, Monk D
Chromatin immunoprecipitation indirect peaks highlight long-range interactions of insulator proteins and Pol II pausing.
Publié le 20 Fév 2014 dans Molecular cell , vol. 53 - pp 672-81
Liang J, Lacroix L, Gamot A, Cuddapah S, Queille S, Lhoumaud P, Lepetit P, Martin PG, Vogelmann J, Court F , Hennion M, Micas G, Urbach S, Bouchez O, Nöllmann M, Zhao K, Emberly E, Cuvier O
Hypermethylation of the alternative AWT1 promoter in hematological malignancies is a highly specific marker for acute myeloid leukemias despite high expression levels.
Publié le 09 Jan 2014 dans Journal of hematology & oncology , vol. 7 - pp 4
Guillaumet-Adkins A, Richter J, Odero MD, Sandoval J, Agirre X, Catala A, Esteller M, Prósper F, Calasanz MJ, Buño I, Kwon M, Court F , Siebert R, Monk D
Comparison of three methods of diagnosis of plasma unmeasured anions in critically ill patients.
Publié le 30 Oct 2013 dans Minerva anestesiologica , vol. 79 - pp 1164-72
Lautrette A, Fejjal M, Aithssain A, Phan TN, Caillot N, Fogli A , Souweine B
Chromatin loop organization of the junb locus in mouse dendritic cells.
Publié le 30 Oct 2013 dans Nucleic acids research , vol. 41 - pp 8908-25
Salem T, Gomard T, Court F , Moquet-Torcy G, Brockly F, Forné T, Piechaczyk M
Stability of genomic imprinting and gestational-age dynamic methylation in complicated pregnancies conceived following assisted reproductive technologies.
Publié le 30 Sep 2013 dans Biology of reproduction , vol. 89 - pp 50
Camprubí C, Iglesias-Platas I, Martin-Trujillo A, Salvador-Alarcon C, Rodriguez MA, Barredo DR, Court F , Monk D
Liver x receptors protect from development of prostatic intra-epithelial neoplasia in mice.
Publié le 30 Mai 2013 dans PLoS genetics , vol. 9 - pp e1003483
Pommier AJ, Dufour J , Alves G, Viennois E, De Boussac H , Trousson A , Volle DH , Caira F , Val P , Arnaud P , Lobaccaro JM , Baron S
Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes.
Publié le 30 Avr 2013 dans Human mutation , vol. 34 - pp 595-602
Court F , Martin-Trujillo A, Romanelli V, Garin I, Iglesias-Platas I, Salafsky I, Guitart M, Perez de Nanclares G, Lapunzina P, Monk D
Myod and H19-Igf2 locus interactions are required for diaphragm formation in the mouse.
Publié le 30 Mar 2013 dans Development (Cambridge, England) , vol. 140 - pp 1231-9
Borensztein M, Monnier P, Court F , Louault Y, Ripoche MA, Tiret L, Yao Z, Tapscott SJ, Forné T, Montarras D, Dandolo L
Imprinting at the PLAGL1 domain is contained within a 70-kb CTCF/cohesin-mediated non-allelic chromatin loop.
Publié le 01 Fév 2013 dans Nucleic acids research , vol. 41 - pp 2171-9
Iglesias-Platas I, Court F , Camprubi C, Sparago A, Guillaumet-Adkins A, Martin-Trujillo A, Riccio A, Moore GE, Monk D
A yeast purification system for human translation initiation factors eIF2 and eIF2Bε and their use in the diagnosis of CACH/VWM disease.
Publié le 01 Jan 2013 dans PloS one , vol. 8 - pp e53958
de Almeida RA, Fogli A , Gaillard M, Scheper GC, Boesflug-Tanguy O, Pavitt GD
Characterization of novel paternal ncRNAs at the Plagl1 locus, including Hymai, predicted to interact with regulators of active chromatin.
Publié le 19 Juin 2012 dans PloS one , vol. 7 - pp e38907
Iglesias-Platas I, Martin-Trujillo A, Cirillo D, Court F , Guillaumet-Adkins A, Camprubi C, Bourc'his D, Hata K, Feil R, Tartaglia G, Arnaud P , Monk D
Transcription and histone methylation changes correlate with imprint acquisition in male germ cells.
Publié le 01 Fév 2012 dans The EMBO journal , vol. 31 - pp 606-15
Henckel A, Chebli K, Kota SK, Arnaud P , Feil R
Relevance of GJC2 promoter mutation in Pelizaeus-Merzbacher-like disease.
Publié le 30 Jan 2012 dans Annals of neurology , vol. 71 - pp 146-8
Combes P, Kammoun N, Monnier A, Gonthier-Guéret C, Giraud G , Bertini E, Chahnez T, Fakhfakh F, Boespflug-Tanguy O, Vaurs-Barrière C
Sjögren-Larsson syndrome: novel mutations in the ALDH3A2 gene in a French cohort.
Publié le 15 Jan 2012 dans Journal of the neurological sciences , vol. 312 - pp 123-6
Sarret C, Rigal M , Vaurs-Barrière C , Dorboz I, Eymard-Pierre E, Combes P, Giraud G , Wanders RJ, Afenjar A, Francannet C, Boespflug-Tanguy O
H19 antisense RNA can up-regulate Igf2 transcription by activation of a novel promoter in mouse myoblasts.
Publié le 01 Jan 2012 dans PloS one , vol. 7 - pp e37923
Tran VG, Court F , Duputié A, Antoine E, Aptel N, Milligan L, Carbonell F, Lelay-Taha MN, Piette J, Weber M, Montarras D, Pinset C, Dandolo L, Forné T, Cathala G
CSF N-glycan profiles to investigate biomarkers in brain developmental disorders: application to leukodystrophies related to eIF2B mutations.
Publié le 01 Jan 2012 dans PloS one , vol. 7 - pp e42688
Fogli A , Merle C, Roussel V, Schiffmann R, Ughetto S, Theisen M, Boespflug-Tanguy O
Developmental splicing deregulation in leukodystrophies related to EIF2B mutations.
Publié le 01 Jan 2012 dans PloS one , vol. 7 - pp e38264
Huyghe A, Horzinski L, Hénaut A, Gaillard M, Bertini E, Schiffmann R, Rodriguez D, Dantal Y, Boespflug-Tanguy O, Fogli A
Characterization of novel paternal ncRNAs at the Plagl1 locus, including Hymai, predicted to interact with regulators of active chromatin.
Publié le 01 Jan 2012 dans PloS one , vol. 7 - pp e38907
Iglesias-Platas I, Martin-Trujillo A, Cirillo D, Court F , Guillaumet-Adkins A, Camprubi C, Bourc'his D, Hata K, Feil R, Tartaglia G, Arnaud P , Monk D
Corneal transduction by intra-stromal injection of AAV vectors in vivo in the mouse and ex vivo in human explants.
Publié le 01 Jan 2012 dans PloS one , vol. 7 - pp e35318
Hippert C, Ibanes S, Serratrice N, Court F , Malecaze F, Kremer EJ, Kalatzis V
[Natural history of adult-onset eIF2B-related disorders: a multicentric survey of 24 cases].
Publié le 30 Nov 2011 dans Revue neurologique , vol. 167 - pp 802-11
Carra-Dalliere C, Horzinski L, Ayrignac X, Vukusic S, Rodriguez D, Mauguiere F, Peter L, Goizet C, Bouhour F, Denier C, Confavreux C, Obadia M, Blanc F, de Seze J, Sedel F, Guennoc AM, Sartori E, Laplaud D, Antoine JC, Fogli A , Boespflug-Tanguy O, Labauge P
Synergic reprogramming of mammalian cells by combined exposure to mitotic Xenopus egg extracts and transcription factors.
Publié le 18 Oct 2011 dans Proceedings of the National Academy of Sciences of the United States of America , vol. 108 - pp 17331-6
Ganier O, Bocquet S, Peiffer I, Brochard V, Arnaud P , Puy A, Jouneau A, Feil R, Renard JP, Méchali M
Long-range chromatin interactions at the mouse Igf2/H19 locus reveal a novel paternally expressed long non-coding RNA.
Publié le 30 Août 2011 dans Nucleic acids research , vol. 39 - pp 5893-906
Court F , Baniol M, Hagege H, Petit JS, Lelay-Taha MN, Carbonell F, Weber M, Cathala G, Forne T
Human imprinted retrogenes exhibit non-canonical imprint chromatin signatures and reside in non-imprinted host genes.
Publié le 30 Juin 2011 dans Nucleic acids research , vol. 39 - pp 4577-86
Monk D, Arnaud P , Frost JM, Wood AJ, Cowley M, Martin-Trujillo A, Guillaumet-Adkins A, Iglesias Platas I, Camprubi C, Bourc'his D, Feil R, Moore GE, Oakey RJ
Molecular genetic analysis of the PLP1 gene in 38 families with PLP1-related disorders: identification and functional characterization of 11 novel PLP1 mutations.
Publié le 16 Juin 2011 dans Orphanet journal of rare diseases , vol. 6 - pp 40
Grossi S, Regis S, Biancheri R, Mort M, Lualdi S, Bertini E, Uziel G, Boespflug-Tanguy O, Simonati A, Corsolini F, Demir E, Marchiani V, Percesepe A, Stanzial F, Rossi A, Vaurs-Barrière C , Cooper DN, Filocamo M
Neurodegenerative disorder related to AIMP1/p43 mutation is not a PMLD.
Publié le 11 Mar 2011 dans American journal of human genetics , vol. 88 - pp 392-3; author reply 393-5
Boespflug-Tanguy O, Aubourg P, Dorboz I, Bégou M, Giraud G , Sarret C, Vaurs-Barrière C
Modulated contact frequencies at gene-rich loci support a statistical helix model for mammalian chromatin organization.
Publié le 01 Jan 2011 dans Genome biology , vol. 12 - pp R42
Court F , Miro J, Braem C, Lelay-Taha MN, Brisebarre A, Atger F, Gostan T, Weber M, Cathala G, Forné T
[Coupling proteinemia and serum protein electrophoresis: evaluation of the capillary technique (Capillarys 2, Sebia), experience from Clermont-Ferrand].
Publié le 01 Nov 2010 dans Annales de biologie clinique , vol. 68 - pp 657-67
Evaluation of the endoplasmic reticulum-stress response in eIF2B-mutated lymphocytes and lymphoblasts from CACH/VWM patients.
Publié le 19 Oct 2010 dans BMC neurology , vol. 10 - pp 94
Horzinski L, Kantor L, Huyghe A, Schiffmann R, Elroy-Stein O, Boespflug-Tanguy O, Fogli A
Genomic imprinting in germ cells: imprints are under control.
Publié le 30 Sep 2010 dans Reproduction (Cambridge, England) , vol. 140 - pp 411-23
Ring1B and Suv39h1 delineate distinct chromatin states at bivalent genes during early mouse lineage commitment.
Publié le 01 Août 2010 dans Development (Cambridge, England) , vol. 137 - pp 2483-92
Alder O, Lavial F, Helness A, Brookes E, Pinho S, Chandrashekran A, Arnaud P , Pombo A, O'Neill L, Azuara V
Genome-wide identification of new imprinted genes.
Publié le 30 Juil 2010 dans Briefings in functional genomics , vol. 9 - pp 304-14
Henckel A, Arnaud P
Peptidomics analysis of lymphoblastoid cell lines.
Publié le 01 Jan 2010 dans Methods in molecular biology (Clifton, N.J.) , vol. 615 - pp 247-57
Fogli A , Bulet P
Eukaryotic initiation factor 2B (eIF2B) GEF activity as a diagnostic tool for EIF2B-related disorders.
Publié le 15 Déc 2009 dans PloS one , vol. 4 - pp e8318
Horzinski L, Huyghe A, Cardoso MC, Gonthier C , Ouchchane L, Schiffmann R, Blanc P , Boespflug-Tanguy O, Fogli A
Histone methylation is mechanistically linked to DNA methylation at imprinting control regions in mammals.
Publié le 15 Sep 2009 dans Human molecular genetics , vol. 18 - pp 3375-83
Henckel A, Nakabayashi K, Sanz LA, Feil R, Hata K, Arnaud P
Natural history of adult-onset eIF2B-related disorders: a multi-centric survey of 16 cases.
Publié le 30 Août 2009 dans Brain : a journal of neurology , vol. 132 - pp 2161-9
Labauge P, Horzinski L, Ayrignac X, Blanc P , Vukusic S, Rodriguez D, Mauguiere F, Peter L, Goizet C, Bouhour F, Denier C, Confavreux C, Obadia M, Blanc F, de Sèze J, Fogli A , Boespflug-Tanguy O
Reciprocal imprinting of human GRB10 in placental trophoblast and brain: evolutionary conservation of reversed allelic expression.
Publié le 15 Août 2009 dans Human molecular genetics , vol. 18 - pp 3066-74
Monk D, Arnaud P , Frost J, Hills FA, Stanier P, Feil R, Moore GE
Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects.
Publié le 30 Jan 2009 dans Annals of neurology , vol. 65 - pp 114-8
Vaurs-Barrière C , Deville M, Sarret C, Giraud G , Des Portes V, Prats-Viñas JM, De Michele G, Dan B, Brady AF, Boespflug-Tanguy O, Touraine R
A mono-allelic bivalent chromatin domain controls tissue-specific imprinting at Grb10.
Publié le 08 Oct 2008 dans The EMBO journal , vol. 27 - pp 2523-32
Sanz LA, Chamberlain S, Sabourin JC, Henckel A, Magnuson T, Hugnot JP, Feil R, Arnaud P
PLP1 splicing abnormalities identified in Pelizaeus-Merzbacher disease and SPG2 fibroblasts are associated with different types of mutations.
Publié le 30 Août 2008 dans Human mutation , vol. 29 - pp 1028-36
Bonnet-Dupeyron MN, Combes P, Santander P, Cailloux F, Boespflug-Tanguy O, Vaurs-Barrière C
Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing.
Publié le 30 Août 2008 dans Genome research , vol. 18 - pp 1270-81
Monk D, Wagschal A, Arnaud P , Müller PS, Parker-Katiraee L, Bourc'his D, Scherer SW, Feil R, Stanier P, Moore GE
Absence of OLIG2 mutations in patients presenting with a severe Pelizaeus-Merzbacher-like leukodystrophy associated with motor neuron dysfunction.
Publié le 05 Juin 2008 dans American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics , vol. 147B - pp 538-9
Bonnet-Dupeyron MN, Combes P, Boespflug-Tanguy O, Vaurs-Barrière C
Sensitivity and specificity of decreased CSF asialotransferrin for eIF2B-related disorder.
Publié le 03 Juin 2008 dans Neurology , vol. 70 - pp 2226-32
Vanderver A, Hathout Y, Maletkovic J, Gordon ES, Mintz M, Timmons M, Hoffman EP, Horzinski L, Niel F, Fogli A , Boespflug-Tanguy O, Schiffmann R
Genes involved in leukodystrophies: a glance at glial functions.
Publié le 30 Mai 2008 dans Current neurology and neuroscience reports , vol. 8 - pp 217-29
Boespflug-Tanguy O, Labauge P, Fogli A , Vaurs-Barriere C
No evidence for association between the EIF2B5 gene and multiple sclerosis in French families.
Publié le 30 Mai 2008 dans Multiple sclerosis (Houndmills, Basingstoke, England) , vol. 14 - pp 573
Fogli A , Barbier C, Cournu-Rebeix I, Babron MC, Clerget-Darpoux F, Fontaine B, Boespflug-Tanguy O
Exon deletion in the non-catalytic domain of eIF2Bepsilon due to a splice site mutation leads to infantile forms of CACH/VWM with severe decrease of eIF2B GEF activity.
Publié le 30 Mai 2008 dans Annals of human genetics , vol. 72 - pp 410-5
Horzinski L, Gonthier C , Rodriguez D, Scherer C, Boespflug-Tanguy O, Fogli A